Bra podcast
Sveriges mest populära poddar
- Affärsnyheter
- Alternativ hälsa
- Amerikansk fotboll
- Andlighet
- Animering och manga
- Astronomi
- Barn och familj
- Baseball
- Basket
- Berättelser för barn
- Böcker
- Brottning
- Buddhism
- Dagliga nyheter
- Dans och teater
- Design
- Djur
- Dokumentär
- Drama
- Efterprogram
- Entreprenörskap
- Fantasysporter
- Filmhistoria
- Filmintervjuer
- Filmrecensioner
- Filosofi
- Flyg
- Föräldraskap
- Fordon
- Fotboll
- Fritid
- Fysik
- Geovetenskap
- Golf
- Hälsa och motion
- Hantverk
- Hinduism
- Historia
- Hobbies
- Hockey
- Hus och trädgård
- Ideell
- Improvisering
- Investering
- Islam
- Judendom
- Karriär
- Kemi
- Komedi
- Komedifiktion
- Komediintervjuer
- Konst
- Kristendom
- Kurser
- Ledarskap
- Life Science
- Löpning
- Marknadsföring
- Mat
- Matematik
- Medicin
- Mental hälsa
- Mode och skönhet
- Motion
- Musik
- Musikhistoria
- Musikintervjuer
- Musikkommentarer
- Näringslära
- Näringsliv
- Natur
- Naturvetenskap
- Nyheter
- Nyhetskommentarer
- Personliga dagböcker
- Platser och resor
- Poddar
- Politik
- Relationer
- Religion
- Religion och spiritualitet
- Rugby
- Så gör man
- Sällskapsspel
- Samhälle och kultur
- Samhällsvetenskap
- Science fiction
- Sexualitet
- Simning
- Självhjälp
- Skönlitteratur
- Spel
- Sport
- Sportnyheter
- Språkkurs
- Stat och kommun
- Ståupp
- Tekniknyheter
- Teknologi
- Tennis
- TV och film
- TV-recensioner
- Underhållningsnyheter
- Utbildning
- Utbildning för barn
- Verkliga brott
- Vetenskap
- Vildmarken
- Visuell konst
Start / the bioinformatics chat
the bioinformatics chat
A podcast about computational biology, bioinformatics, and next generation sequencing.
70 avsnitt • Längd: 65 min • Oregelbundet
Om podden
A podcast about computational biology, bioinformatics, and next generation sequencing.
The podcast the bioinformatics chat is created by Roman Cheplyaka. The podcast and the artwork on this page are embedded on this page using the public podcast feed (RSS).
Avsnitt
#70 Prioritizing drug target genes with Marie Sadler
21 december 2023 |
52 min
In this episode, Marie Sadler talks about her recent Cell Genomics paper, Multi-layered genetic approaches to identify approved drug targets.
Previous studies have found that the drugs that target a gene linked to the disease are more likely to be approved. Yet there are many ways to define what it means for a gene to be linked to the disease. Perhaps the most straightforward approach is to rely on the genome-wide association studies (GWAS) data, but that data can also be integrated with quantitative trait loci (eQTL or pQTL) information to establish less obvious links between genetic variants (which often lie outside of genes) and genes. Finally, there’s exome sequencing, which, unlike GWAS, captures rare genetic variants. So in this paper, Marie and her colleagues set out to benchmark these different methods against one another.
Listen to the episode to find out how these methods work, which ones work better, and how network propagation can improve the prediction accuracy.
Links:
- Multi-layered genetic approaches to identify approved drug targets (Marie C. Sadler, Chiara Auwerx, Patrick Deelen, Zoltán Kutalik)
- Marie on GitHub
- Interview with Mariana Mamonova, the Ukrainian marine infantry combat medic who spent 6 months in russian captivity while pregnant
Thank you to Jake Yeung, Michael Weinstein, and other Patreon members for supporting this episode.
#69 Suffix arrays in optimal compressed space and δ-SA with Tomasz Kociumaka and Dominik Kempa
29 september 2023 |
57 min
Today on the podcast we have Tomasz Kociumaka and Dominik Kempa, the authors of the preprint Collapsing the Hierarchy of Compressed Data Structures: Suffix Arrays in Optimal Compressed Space.
The suffix array is one of the foundational data structures in bioinformatics, serving as an index that allows fast substring searches in a large text. However, in its raw form, the suffix array occupies the space proportional to (and several times larger than) the original text.
In their paper, Tomasz and Dominik construct a new index, δ-SA, which on the one hand can be used in the same way (answer the same queries) as the suffix array and the inverse suffix array, and on the other hand, occupies the space roughly proportional to the gzip’ed text (or, more precisely, to the measure δ that they define — hence the name).
Moreover, they mathematically prove that this index is optimal, in the sense that any index that supports these queries — or even much weaker queries, such as simply accessing the i-th character of the text — cannot be significantly smaller (as a function of δ) than δ-SA.
Links:
- Collapsing the Hierarchy of Compressed Data Structures: Suffix Arrays in Optimal Compressed Space (Dominik Kempa, Tomasz Kociumaka)
Thank you to Jake Yeung and other Patreon members for supporting this episode.
#68 Phylogenetic inference from raw reads and Read2Tree with David Dylus
28 augusti 2023 |
49 min
In this episode, David Dylus talks about Read2Tree, a tool that builds alignment matrices and phylogenetic trees from raw sequencing reads. By leveraging the database of orthologous genes called OMA, Read2Tree bypasses traditional, time-consuming steps such as genome assembly, annotation and all-versus-all sequence comparisons.
Links:
- Inference of phylogenetic trees directly from raw sequencing reads using Read2Tree (David Dylus, Adrian Altenhoff, Sina Majidian, Fritz J. Sedlazeck, Christophe Dessimoz)
- Background story
- Read2Tree on GitHub
- OMA browser
- The Guardian’s podcast about Victoria Amelina and Volodymyr Vakulenko
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#67 AlphaFold and variant effect prediction with Amelie Stein
29 juli 2023 |
35 min
This is the third and final episode in the AlphaFold series, originally recorded on February 23, 2022, with Amelie Stein, now an associate professor at the University of Copenhagen.
In the episode, Amelie explains what 𝛥𝛥G is, how it informs us whether a particular protein mutation affects its stability, and how AlphaFold 2 helps in this analysis.
A note from Amelie:
Something that has happened in the meantime is the publication of methods that predict 𝛥𝛥G with ML methods, so much faster than Rosetta. One of them, RaSP, is from our group, while ddMut is from another subset of authors of the AF2 community assessment paper.
Other links:
- A structural biology community assessment of AlphaFold2 applications (Mehmet Akdel, Douglas E. V. Pires, Eduard Porta Pardo, Jürgen Jänes, Arthur O. Zalevsky, Bálint Mészáros, Patrick Bryant, Lydia L. Good, Roman A. Laskowski, Gabriele Pozzati, Aditi Shenoy, Wensi Zhu, Petras Kundrotas, Victoria Ruiz Serra, Carlos H. M. Rodrigues, Alistair S. Dunham, David Burke, Neera Borkakoti, Sameer Velankar, Adam Frost, Jérôme Basquin, Kresten Lindorff-Larsen, Alex Bateman, Andrey V. Kajava, Alfonso Valencia, Sergey Ovchinnikov, Janani Durairaj, David B. Ascher, Janet M. Thornton, Norman E. Davey, Amelie Stein, Arne Elofsson, Tristan I. Croll & Pedro Beltrao)
- A crime in the making: Russia’s atrocities — the podcast episode about the Olenivka prison massacre
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#66 AlphaFold and shape-mers with Janani Durairaj
10 juli 2023 |
21 min
This is the second episode in the AlphaFold series, originally recorded on February 14, 2022, with Janani Durairaj, a postdoctoral researcher at the University of Basel.
Janani talks about how she used shape-mers and topic modelling to discover classes of proteins assembled by AlphaFold 2 that were absent from the Protein Data Bank (PDB).
The bioinformatics discussion starts at 03:35.
Links:
- A structural biology community assessment of AlphaFold2 applications (Mehmet Akdel, Douglas E. V. Pires, Eduard Porta Pardo, Jürgen Jänes, Arthur O. Zalevsky, Bálint Mészáros, Patrick Bryant, Lydia L. Good, Roman A. Laskowski, Gabriele Pozzati, Aditi Shenoy, Wensi Zhu, Petras Kundrotas, Victoria Ruiz Serra, Carlos H. M. Rodrigues, Alistair S. Dunham, David Burke, Neera Borkakoti, Sameer Velankar, Adam Frost, Jérôme Basquin, Kresten Lindorff-Larsen, Alex Bateman, Andrey V. Kajava, Alfonso Valencia, Sergey Ovchinnikov, Janani Durairaj, David B. Ascher, Janet M. Thornton, Norman E. Davey, Amelie Stein, Arne Elofsson, Tristan I. Croll & Pedro Beltrao)
- The Protein Universe Atlas
- What is hidden in the darkness? Deep-learning assisted large-scale protein family curation uncovers novel protein families and folds (Janani Durairaj, Andrew M. Waterhouse, Toomas Mets, Tetiana Brodiazhenko, Minhal Abdullah, Gabriel Studer, Mehmet Akdel, Antonina Andreeva, Alex Bateman, Tanel Tenson, Vasili Hauryliuk, Torsten Schwede, Joana Pereira)
- Geometricus: Protein Structures as Shape-mers derived from Moment Invariants on GitHub
- The group page
- The Folded Weekly newsletter
- A New York Times article about the Kramatorsk missile strike. The Instagram video, part of which you can hear at the beginning of the episode, appears to have been deleted.
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#65 AlphaFold and protein interactions with Pedro Beltrao
21 juni 2023 |
52 min
In this episode, originally recorded on February 9, 2022, Roman talks to Pedro Beltrao about AlphaFold, the software developed by DeepMind that predicts a protein’s 3D structure from its amino acid sequence.
Pedro is an associate professor at ETH Zurich and the coordinator of the structural biology community assessment of AlphaFold2 applications project, which involved over 30 scientists from different institutions.
Pedro talks about the origins of the project, its main findings, the importance of the confidence metric that AlphaFold assigns to its predictions, and Pedro’s own area of interest — predicting pockets in proteins and protein-protein interactions.
Links:
- A structural biology community assessment of AlphaFold2 applications (Mehmet Akdel, Douglas E. V. Pires, Eduard Porta Pardo, Jürgen Jänes, Arthur O. Zalevsky, Bálint Mészáros, Patrick Bryant, Lydia L. Good, Roman A. Laskowski, Gabriele Pozzati, Aditi Shenoy, Wensi Zhu, Petras Kundrotas, Victoria Ruiz Serra, Carlos H. M. Rodrigues, Alistair S. Dunham, David Burke, Neera Borkakoti, Sameer Velankar, Adam Frost, Jérôme Basquin, Kresten Lindorff-Larsen, Alex Bateman, Andrey V. Kajava, Alfonso Valencia, Sergey Ovchinnikov, Janani Durairaj, David B. Ascher, Janet M. Thornton, Norman E. Davey, Amelie Stein, Arne Elofsson, Tristan I. Croll & Pedro Beltrao)
- Pedro’s group at ETH Zurich
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#64 Enformer: predicting gene expression from sequence with Žiga Avsec
9 november 2021 |
60 min
In this episode, Jacob Schreiber interviews Žiga Avsec about a recently released model, Enformer. Their discussion begins with life differences between academia and industry, specifically about how research is conducted in the two settings. Then, they discuss the Enformer model, how it builds on previous work, and the potential that models like it have for genomics research in the future. Finally, they have a high-level discussion on the state of modern deep learning libraries and which ones they use in their day-to-day developing.
Links:
- Effective gene expression prediction from sequence by integrating long-range interactions (Žiga Avsec, Vikram Agarwal, Daniel Visentin, Joseph R. Ledsam, Agnieszka Grabska-Barwinska, Kyle R. Taylor, Yannis Assael, John Jumper, Pushmeet Kohli & David R. Kelley )
- DeepMind Blog Post (Žiga Avsec)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#63 Bioinformatics Contest 2021 with Maksym Kovalchuk and James Matthew Holt
27 september 2021 |
61 min
The Bioinformatics Contest is back this year, and we are back to discuss it!
This year’s contest winners Maksym Kovalchuk (1st prize) and Matt Holt (2nd prize) talk about how they approach participating in the contest and what strategies have earned them the top scores.
Timestamps and links for the individual problems:
- 00:10:36 Genotype Imputation
- 00:21:26 Causative Mutation
- 00:30:27 Superspreaders
- 00:37:22 Minor Haplotype
- 00:46:37 Isoform Matching
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#62 Steady states of metabolic networks and Dingo with Apostolos Chalkis
28 juli 2021 |
38 min
In this episode, Apostolos Chalkis presents sampling steady states of metabolic networks as an alternative to the widely used flux balance analysis (FBA). We also discuss dingo, a Python package written by Apostolos that employs geometric random walks to sample steady states. You can see dingo in action here.
Links:
- Dingo on GitHub
- Searching for COVID-19 treatments using metabolic networks
- Tweag open source fellowships
- This episode was originally published on the Compositional podcast.
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#61 3D genome organization and GRiNCH with Da-Inn Erika Lee
23 juni 2021 |
70 min
In this episode, Jacob Schreiber interviews Da-Inn Erika Lee about data and computational methods for making sense of 3D genome structure. They begin their discussion by talking about 3D genome structure at a high level and the challenges in working with such data. Then, they discuss a method recently developed by Erika, named GRiNCH, that mines this data to identify spans of the genome that cluster together in 3D space and potentially help control gene regulation.
Links:
- GRiNCH: simultaneous smoothing and detection of topological units of genome organization from sparse chromatin contact count matrices with matrix factorization (Da-Inn Lee and Sushmita Roy)
- GRiNCH Project Page
- In silico prediction of high-resolution Hi-C interaction matrices (Shilu Zhang, Deborah Chasman, Sara Knaack, and Sushmita Roy)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#60 Differential gene expression and DESeq2 with Michael Love
12 maj 2021 |
91 min
In this episode, Michael Love joins us to talk about the differential gene expression analysis from bulk RNA-Seq data.
We talk about the history of Mike’s own differential expression package, DESeq2, as well as other packages in this space, like edgeR and limma, and the theory they are based upon. Mike also shares his experience of being the author and maintainer of a popular bioninformatics package.
Links:
- Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 (Love, M.I., Huber, W. & Anders, S.)
- DESeq2 on Bioconductor
- Chan Zuckerberg Initiative: Ensuring Reproducible Transcriptomic Analysis with DESeq2 and tximeta
And a more comprehensive set of links from Mike himself:
limma, the original paper and limma-voom:
https://pubmed.ncbi.nlm.nih.gov/16646809/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4053721/
edgeR papers:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2796818/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3378882/
The recent manuscript mentioned from the Kendziorski lab, which has a Gamma-Poisson hierarchical structure, although it does not in general reduce to the Negative Binomial:
https://doi.org/10.1101/2020.10.28.359901
We talk about robust steps for estimating the middle of the dispersion prior distribution, references are Anders and Huber 2010 (DESeq), Eling et al 2018 (one of the BASiCS papers), and Phipson et al 2016:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3218662/ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6167088/ https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5373812/
The Stan software:
https://mc-stan.org/
We talk about using publicly available data as a prior, references I mention are the McCall et al paper using publicly available data to ask if a gene is expressed, and a new manuscript from my lab that compares splicing in a sample to GTEx as a reference panel:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3013751/ https://doi.org/10.1101/856401
Regarding estimating the width of the dispersion prior, references are the Robinson and Smyth 2007 paper, McCarthy et al 2012 (edgeR), and Wu et al 2013 (DSS):
https://pubmed.ncbi.nlm.nih.gov/17881408/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3378882/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3590927/
Schurch et al 2016, a RNA-seq dataset with many replicates, helpful for benchmarking:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4878611/
Stephens paper on the false sign rate (ash):
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5379932/
Heavy-tailed distributions for effect sizes, Zhu et al 2018:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6581436/
I credit Kevin Blighe and Alexander Toenges, who help to answer lots of DESeq2 questions on the support site:
https://www.biostars.org/u/41557/
https://www.biostars.org/u/25721/
The EOSS award, which has funded vizWithSCE by Kwame Forbes, and nullranges by Wancen Mu and Eric Davis:
https://chanzuckerberg.com/eoss/proposals/ensuring-reproducible-transcriptomic-analysis-with-deseq2-and-tximeta/
https://kwameforbes.github.io/vizWithSCE/
https://nullranges.github.io/nullranges/
One of the recent papers from my lab, MRLocus for eQTL and GWAS integration:
https://mikelove.github.io/mrlocus/
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#59 Proteomics calibration with Lindsay Pino
21 april 2021 |
48 min
In this episode, Lindsay Pino discusses the challenges of making quantitative measurements in the field of proteomics. Specifically, she discusses the difficulties of comparing measurements across different samples, potentially acquired in different labs, as well as a method she has developed recently for calibrating these measurements without the need for expensive reagents. The discussion then turns more broadly to questions in genomics that can potentially be addressed using proteomic measurements.
Links:
- Talus Bioscience
- Matrix-Matched Calibration Curves for Asssessing Analytical Figures of Merit in Quantitative Proteomics (Lindsay K. Pino, Brian C. Searle, Han-Yin Yang, Andrew N. Hoofnagle, William S. Noble, and Michael J. MacCross)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#58 B cell maturation and class switching with Hamish King
31 mars 2021 |
89 min
In this episode, we learn about B cell maturation and class switching from Hamish King. Hamish recently published a paper on this subject in Science Immunology, where he and his coauthors analyzed gene expression and antibody repertoire data from human tonsils. In the episode Hamish talks about some of the interesting B cell states he uncovered and shares his thoughts on questions such as «When does a B cell decide to class-switch?» and «Why is the antibody isotype correlated with its affinity?»
Links:
- Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics (Hamish W. King, Nara Orban, John C. Riches, Andrew J. Clear, Gary Warnes, Sarah A. Teichmann, Louisa K. James) (paywalled by Science Immunology)
- Antibody repertoire and gene expression dynamics of diverse human B cell states during affinity maturation (the preprint of the above Science Immunology paper)
- www.tonsilimmune.org: An immune cell atlas of the human tonsil and B cell maturation
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#57 Enhancers with Molly Gasperini
10 mars 2021 |
47 min
In this episode, Jacob Schreiber interviews Molly Gasperini about enhancer elements. They begin their discussion by talking about Octant Bio, and then dive into the surprisingly difficult task of defining enhancers and determining the mechanisms that enable them to regulate gene expression.
Links:
- Octant Bio
- Towards a comprehensive catalogue of validated and target-linked human enhancers (Molly Gasperini, Jacob M. Tome, and Jay Shendure)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#56 Polygenic risk scores in admixed populations with Bárbara Bitarello
17 februari 2021 |
90 min
Polygenic risk scores (PRS) rely on the genome-wide association studies (GWAS) to predict the phenotype based on the genotype. However, the prediction accuracy suffers when GWAS from one population are used to calculate PRS within a different population, which is a problem because the majority of the GWAS are done on cohorts of European ancestry.
In this episode, Bárbara Bitarello helps us understand how PRS work and why they don’t transfer well across populations.
Links:
- Polygenic Scores for Height in Admixed Populations (Bárbara D. Bitarello, Iain Mathieson)
- What is ancestry? (Iain Mathieson, Aylwyn Scally)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#55 Phylogenetics and the likelihood gradient with Xiang Ji
13 januari 2021 |
57 min
In this episode, we chat about phylogenetics with Xiang Ji. We start with a general introduction to the field and then go deeper into the likelihood-based methods (maximum likelihood and Bayesian inference). In particular, we talk about the different ways to calculate the likelihood gradient, including a linear-time exact gradient algorithm recently published by Xiang and his colleagues.
Links:
- Gradients Do Grow on Trees: A Linear-Time O(N)-Dimensional Gradient for Statistical Phylogenetics (Xiang Ji, Zhenyu Zhang, Andrew Holbrook, Akihiko Nishimura, Guy Baele, Andrew Rambaut, Philippe Lemey, Marc A Suchard)
- BEAGLE: the package that implements the gradient algorithm
- BEAST: the program that implements the Hamiltonian Monte Carlo sampler and the molecular clock models
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#54 Seeding methods for read alignment with Markus Schmidt
16 december 2020 |
61 min
In this episode, Markus Schmidt explains how seeding in read alignment works. We define and compare k-mers, minimizers, MEMs, SMEMs, and maximal spanning seeds. Markus also presents his recent work on computing variable-sized seeds (MEMs, SMEMs, and maximal spanning seeds) from fixed-sized seeds (k-mers and minimizers) and his Modular Aligner.
Links:
- A performant bridge between fixed-size and variable-size seeding (Arne Kutzner, Pok-Son Kim, Markus Schmidt)
- MA the Modular Aligner
- Calibrating Seed-Based Heuristics to Map Short Reads With Sesame (Guillaume J. Filion, Ruggero Cortini, Eduard Zorita) — another interesting recent work on seeding methods (though we didn’t get to discuss it in this episode)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#53 Real-time quantitative proteomics with Devin Schweppe
18 november 2020 |
63 min
In this episode, Jacob Schreiber interviews Devin Schweppe about the analysis of mass spectrometry data in the field of proteomics. They begin by delving into the different types of mass spectrometry methods, including MS1, MS2, and, MS3, and the reasons for using each. They then discuss a recent paper from Devin, Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics that involved building a real-time system for quantifying proteomic samples from MS3, and the types of analyses that this system allows one to do.
Links:
- Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics (Devin K. Schweppe, Jimmy K. Eng, Qing Yu, Derek Bailey, Ramin Rad, Jose Navarrete-Perea, Edward L. Huttlin, Brian K. Erickson, Joao A. Paulo, and Steven P. Gygi)
- Benchmarking the Orbitrap Tribrid Eclipse for Next Generation Multiplexed Proteomics (Qing Yu, Joao A Paulo, Jose Naverrete-Perea, Graeme C McAlister, Jesse D Canterbury, Derek J Bailey, Aaron M Robitaille, Romain Huguet, Vlad Zabrouskov, Steven P Gygi, Devin K Schweppe)
- Improved Monoisotopic Mass Estimation for Deeper Proteome Coverage (Ramin Rad, Jiaming Li, Julian Mintseris, Jeremy O’Connell, Steven P. Gygi, and Devin K. Schweppe)
- Schweppe Lab Website (Hiring!)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#52 How 23andMe finds identical-by-descent segments with William Freyman
27 oktober 2020 |
43 min
In this episode, Will Freyman talks about identity-by-descent (IBD): how it’s used at 23andMe, and how the templated positional Burrows-Wheeler transform can find IBD segments in the presence of genotyping and phasing errors.
Links:
- Fast and robust identity-by-descent inference with the templated positional Burrows-Wheeler transform (William A. Freyman, Kimberly F. McManus, Suyash S. Shringarpure, Ethan M. Jewett, Katarzyna Bryc, the 23andMe Research Team, Adam Auton)
- 23andMe research
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#51 Basset and Basenji with David Kelley
7 oktober 2020 |
74 min
In this episode, Jacob Schreiber interviews David Kelley about machine learning models that can yield insight into the consequences of mutations on the genome. They begin their discussion by talking about Calico Labs, and then delve into a series of papers that David has written about using models, named Basset and Basenji, that connect genome sequence to functional activity and so can be used to quantify the effect of any mutation.
Links:
- Calico Labs
- Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks (David R. Kelley, Jasper Snoek, and John Rinn)
- Sequential regulatory activity prediction across chromosomes with convolutional neural networks (David R. Kelley, Yakir A. Reshef, Maxwell Bileschi, David Belanger, Cory Y. McLean, and Jaspar Snoek)
- Cross-species regulatory sequence activity prediction (David R. Kelley)
- Basenji GitHub Repo
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#50 ENCODE3 with Jill Moore
10 september 2020 |
56 min
In this episode, Jacob Schreiber interviews Jill Moore about recent research from the ENCODE Project. They begin their discussion with an overview and goals of the ENCODE Project, and then discuss a bundle of papers that were recently published in various Nature journals and the flagship paper, Expanded encyclopaedias of DNA elements in the human and mouse genomes. They conclude their discussion by talking about the challenges with managing a large project as a trainee in a consortium setting.
Links:
- Expanded encyclopaedias of DNA elements in the human and mouse genomes (The ENCODE Project Consortium, Jill E. Moore, […], Zhiping Weng)
- SCREEN
- The ENCODE Portal
- The ENCODE3 Publication Bundle
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#49 Most Permissive Boolean Networks with Loïc Paulevé
19 augusti 2020 |
64 min
In systems biology, Boolean networks are a way to model interactions such as gene regulation or cell signaling. The standard interpretations of Boolean networks are the synchronous, asynchronous, and fully asynchronous semantics.
In this episode, Loïc Paulevé explains how the same Boolean networks can be interpreted in a new, “most permissive” way. Loïc proved mathematically that his semantics can reproduce all behaviors achievable by a compatible quantitative model, whereas the traditional interpretations in general cannot. Furthermore, it turns out that deciding whether a certain state in a Boolean network is reachable can be done much more efficiently in MPBNs than in the traditional interpretations.
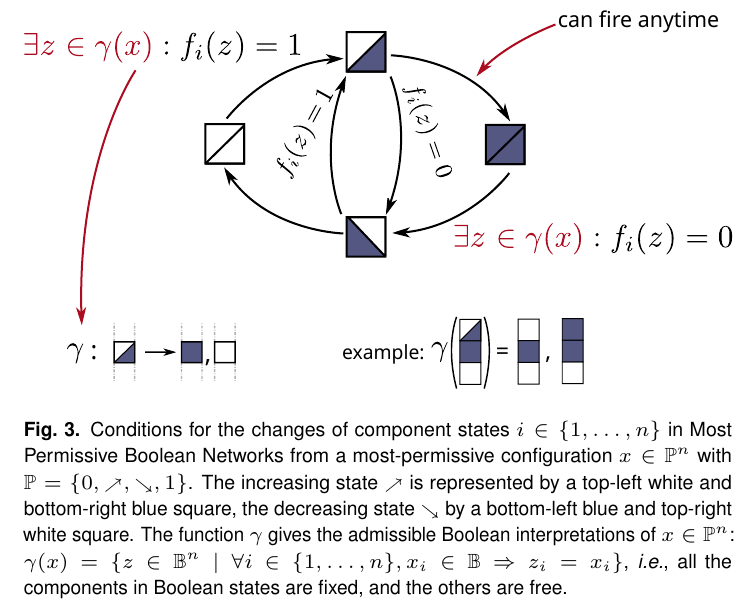
Links:
- Reconciling Qualitative, Abstract, and Scalable Modeling of Biological Networks (Loïc Paulevé, Juraj Kolčák, Thomas Chatain, Stefan Haar)
- mpbn on GitHub: an implementation of reachability and attractor analysis in Most Permissive Boolean Networks
- BoNesis on GitHub: synthesis of Most Permissive Boolean Networks from network architecture and dynamical properties
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#48 Machine learning for drug development with Marinka Zitnik
29 juli 2020 |
85 min
In this episode, Jacob Schreiber interviews Marinka Zitnik about applications of machine learning to drug development. They begin their discussion with an overview of open research questions in the field, including limiting the search space of high-throughput testing methods, designing drugs entirely from scratch, predicting ways that existing drugs can be repurposed, and identifying likely side-effects of combining existing drugs in novel ways. Focusing on the last of these areas, they then discuss one of Marinka’s recent papers, Modeling polypharmacy side effects with graph convolutional networks.
Links:
- Modeling polypharmacy side effects with graph convolutional networks (Marinka Zitnik, Monica Agrawal, Jure Leskovec)
- Network Medicine Framework for Identifying Drug Repurposing Opportunities for COVID-19 (Deisy Morselli Gysi, Ítalo Do Valle, Marinka Zitnik, Asher Ameli, Xiao Gan, Onur Varol, Helia Sanchez, Rebecca Marlene Baron, Dina Ghiassian, Joseph Loscalzo, Albert-László Barabási)
- AI Cures initiative
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#47 Reproducible pipelines and NGLess with Luis Pedro Coelho
24 juni 2020 |
58 min
NGLess is a programming language specifically targeted at next generation sequencing (NGS) data processing. In this episode we chat with its main developer, Luis Pedro Coelho, about the benefits of domain-specific languages, pros and cons of Haskell in bioinformatics, reproducibility, and of course NGLess itself.
Links:
- NGLess on GitHub
- NG-meta-profiler: fast processing of metagenomes using NGLess, a domain-specific language (Luis Pedro Coelho, Renato Alves, Paulo Monteiro, Jaime Huerta-Cepas, Ana Teresa Freitas, Peer Bork)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#46 HiFi reads and HiCanu with Sergey Nurk and Sergey Koren
27 maj 2020 |
69 min
In this episode, I continue to talk (but mostly listen) to Sergey Koren and Sergey Nurk. If you missed the previous episode, you should probably start there. Otherwise, join us to learn about HiFi reads, the tradeoff between read length and quality, and what tricks HiCanu employs to resolve highly similar repeats.
Links:
- HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads (Sergey Nurk, Brian P. Walenz, Arang Rhie, Mitchell R. Vollger, Glennis A. Logsdon, Robert Grothe, Karen H. Miga, Evan E. Eichler, Adam M. Phillippy, Sergey Koren)
- Canu on GitHub (includes the HiCanu mode)
- The Telomere-to-Telomere (T2T) consortium
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#45 Genome assembly and Canu with Sergey Koren and Sergey Nurk
20 maj 2020 |
77 min
In this episode, Sergey Nurk and Sergey Koren from the NIH share their thoughts on genome assembly. The two Sergeys tell the stories behind their amazing careers as well as behind some of the best known genome assemblers: Celera assembler, Canu, and SPAdes.
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#44 DNA tagging and Porcupine with Kathryn Doroschak
29 april 2020 |
45 min
Porcupine is a molecular tagging system—a way to tag physical objects with pieces of DNA called molecular bits, or molbits for short. These DNA tags then can be rapidly sequenced on an Oxford Nanopore MinION device without any need for library preparation.
In this episode, Katie Doroschak explains how Porcupine works—how molbits are designed and prepared, and how they are directly recognized by the software without an intermediate basecalling step.
Links:
- Porcupine: Rapid and robust tagging of physical objects using nanopore-orthogonal DNA strands (Kathryn Doroschak, Karen Zhang, Melissa Queen, Aishwarya Mandyam, Karin Strauss, Luis Ceze, Jeff Nivala)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#43 Generalized PCA for single-cell data with William Townes
27 mars 2020 |
60 min
Will Townes proposes a new, simpler way to analyze scRNA-seq data with unique molecular identifiers (UMIs). Observing that such data is not zero-inflated, Will has designed a PCA-like procedure inspired by generalized linear models (GLMs) that, unlike the standard PCA, takes into account statistical properties of the data and avoids spurious correlations (such as one or more of the top principal components being correlated with the number of non-zero gene counts).
Also check out Will’s paper for a feature selection algorithm based on deviance, which we didn’t get a chance to discuss on the podcast.
Links:
- Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model (F. William Townes, Stephanie C. Hicks, Martin J. Aryee, Rafael A. Irizarry)
- GLM-PCA for R
- GLM-PCA for Python
- scry: an R package for feature selection by deviance (alternative to highly variable genes)
- Droplet scRNA-seq is not zero-inflated (Valentine Svensson)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#42 Spectrum-preserving string sets and simplitigs with Amatur Rahman and Karel Břinda
28 februari 2020 |
53 min
In this episode, we hear from Amatur Rahman and Karel Břinda, who independently of one another released preprints on the same concept, called simplitigs or spectrum-preserving string sets. Simplitigs offer a way to efficiently store and query large sets of k-mers—or, equivalently, large de Bruijn graphs.
Links:
- Simplitigs as an efficient and scalable representation of de Bruijn graphs (Karel Břinda, Michael Baym, Gregory Kucherov)
- Representation of k-mer sets using spectrum-preserving string sets (Amatur Rahman, Paul Medvedev)
- Open mic
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#41 Epidemic models with Kris Parag
27 januari 2020 |
68 min
Kris Parag is here to teach us about the mathematical modeling of infectious disease epidemics. We discuss the SIR model, the renewal models, and how insights from information theory can help us predict where an epidemic is going.
Links:
- Optimising Renewal Models for Real-Time Epidemic Prediction and Estimation (KV Parag, CA Donnelly)
- Adaptive Estimation for Epidemic Renewal and Phylogenetic Skyline Models (KV Parag, CA Donnelly)
- The listener survey
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#40 Plasmid classification and binning with Sergio Arredondo-Alonso and Anita Schürch
30 december 2019 |
45 min
Does a given bacterial gene live on a plasmid or the chromosome? What other genes live on the same plasmid?
In this episode, we hear from Sergio Arredondo-Alonso and Anita Schürch, whose projects mlplasmids and gplas answer these types of questions.
Links:
- mlplasmids: a user-friendly tool to predict plasmid- and chromosome-derived sequences for single species (Sergio Arredondo-Alonso, Malbert R. C. Rogers, Johanna C. Braat, Tess D. Verschuuren, Janetta Top, Jukka Corander, Rob J. L. Willems, Anita C. Schürch)
- gplas: a comprehensive tool for plasmid analysis using short-read graphs (Sergio Arredondo-Alonso, Martin Bootsma, Yaïr Hein, Malbert R.C. Rogers, Jukka Corander, Rob JL Willems, Anita C. Schürch)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#39 Amplicon sequence variants and bias with Benjamin Callahan
29 november 2019 |
62 min
In this episode, Benjamin Callahan talks about some of the issues faced by microbiologists when conducting amplicon sequencing and metagenomic studies. The two main themes are:
- Why one should probably avoid using OTUs (operational taxonomic units) and use exact sequence variants (also called amplicon sequence variants, or ASVs), and how DADA2 manages to deduce the exact sequences present in the sample.
- Why abundances inferred from community sequencing data are biased, and how we can model and correct this bias.
Links:
- Exact sequence variants should replace operational taxonomic units in marker-gene data analysis (Benjamin J Callahan, Paul J McMurdie & Susan P Holmes)
- DADA2: High-resolution sample inference from Illumina amplicon data (Benjamin J Callahan, Paul J McMurdie, Michael J Rosen, Andrew W Han, Amy Jo A Johnson & Susan P Holmes)
- In Nature, There Is Only Diversity (Michael R. McLaren, Benjamin J. Callahan)
- Consistent and correctable bias in metagenomic sequencing experiments (Michael R McLaren, Amy D Willis, Benjamin J Callahan)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#38 Issues in legacy genomes with Luke Anderson-Trocmé
22 oktober 2019 |
61 min
In this episode, Luke Anderson-Trocmé talks about his findings from the 1000 Genomes Project. Namely, the early sequenced genomes sometimes contain specific mutational signatures that haven’t been replicated from other sources and can be found via their association with lower base quality scores. Listen to Luke telling the story of how he stumbled upon and investigated these fake variants and what their impact is.
Links:
- Legacy Data Confounds Genomics Studies (bioRxiv, Molecular Biology and Evolution (paywall)) (Luke Anderson-Trocmé, Rick Farouni, Mathieu Bourgey, Yoichiro Kamatani, Koichiro Higasa, Jeong-Sun Seo, Changhoon Kim, Fumihiko Matsuda and Simon Gravel)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#37 Causality and potential outcomes with Irineo Cabreros
27 september 2019 |
41 min
In this episode, I talk with Irineo Cabreros about causality. We discuss why causality matters, what does and does not imply causality, and two different mathematical formalizations of causality: potential outcomes and directed acyclic graphs (DAGs). Causal models are usually considered external to and separate from statistical models, whereas Irineo’s new paper shows how causality can be viewed as a relationship between particularly chosen random variables (potential outcomes).
Links:
- Causal models on probability spaces (Irineo Cabreros, John D. Storey)
- The Book of Why: The New Science of Cause and Effect (Judea Pearl, Dana Mackenzie)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#36 scVI with Romain Lopez and Gabriel Misrachi
30 augusti 2019 |
80 min
In this episode, we hear from Romain Lopez and Gabriel Misrachi about scVI—Single-cell Variational Inference. scVI is a probabilistic model for single-cell gene expression data that combines a hierarchical Bayesian model with deep neural networks encoding the conditional distributions. scVI scales to over one million cells and can be used for scRNA-seq normalization and batch effect removal, dimensionality reduction, visualization, and differential expression. We also discuss the recently implemented in scVI automatic hyperparameter selection via Bayesian optimization.
Links:
- Deep generative modeling for single-cell transcriptomics (Romain Lopez, Jeffrey Regier, Michael Cole, Michael I. Jordan, Nir Yosef)
- scVI on GitHub
- Should we zero-inflate scVI?
- Hyperparameter search for scVI
- Droplet scRNA-seq is not zero inflated (Valentine Svensson)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#35 The role of the DNA shape in transcription factor binding with Hassan Samee
26 juli 2019 |
62 min
Even though the double-stranded DNA has the famous regular helical shape, there are small variations in the geometry of the helix depending on what exact nucleotides its made of at that position.
In this episode of the bioinformatics chat, Hassan Samee talks about the role the DNA shape plays in recognition of the DNA by DNA-binding proteins, such as transcription factors. Hassan also explains how his algorithm, ShapeMF, can deduce the DNA shape motifs from the ChIP-seq data.
Links:
- A De Novo Shape Motif Discovery Algorithm Reveals Preferences of Transcription Factors for DNA Shape Beyond Sequence Motifs (Md. Abul Hassan Samee, Benoit G. Bruneau, Katherine Pollard)
- ShapeMF on GitHub
- A picture explaining some of the DNA shape features
{kind=link}
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#34 Power laws and T-cell receptors with Kristina Grigaityte
29 juni 2019 |
87 min
An αβ T-cell receptor is composed of two highly variable protein chains, the α chain and the β chain. However, based only on bulk DNA or RNA sequencing it is impossible to determine which of the α chain and β chain sequences were paired in the same receptor.
In this episode, Kristina Grigaityte talks about her analysis of 200,000 paired αβ sequences, which have been obtained by targeted single-cell RNA sequencing. Kristina used the power law distribution to model the T-cell clone sizes, which led her to reject the commonly held assumptions about the independence of the α and β chains. We also talk about Bayesian inference of power law distributions and about mixtures of power laws.
Links:
- Single-cell sequencing reveals αβ chain pairing shapes the T cell repertoire. Kristina Grigaityte, Jason A. Carter, Stephen J. Goldfless, Eric W. Jeffery, Ronald J. Hause, Yue Jiang, David Koppstein, Adrian W. Briggs, George M. Church, Francois Vigneault, Gurinder S. Atwal
- Bayesian inference of power law distributions. Kristina Grigaityte, Gurinder Atwal
- Mathematics in modern immunology. Castro M, Lythe G, Molina-París C, Ribeiro RM.
- Power laws, Pareto distributions and Zipf’s law. M. E. J. Newman
- So You Think You Have a Power Law — Well Isn’t That Special?
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#33 Genome assembly from long reads and Flye with Mikhail Kolmogorov
31 maj 2019 |
73 min
Modern genome assembly projects are often based on long reads in an attempt to bridge longer repeats. However, due to the higher error rate of the current long read sequencers, assemblers based on de Bruijn graphs do not work well in this setting, and the approaches that do work are slower.
In this episode, Mikhail Kolmogorov from Pavel Pevzner’s lab joins us to talk about some of the ideas developed in the lab that made it possible to build a de Bruijn-like assembly graph from noisy reads. These ideas are now implemented in the Flye assembler, which performs much faster than the existing long read assemblers without sacrificing the quality of the assembly.
Links:
- Assembly of Long Error-Prone Reads Using Repeat Graphs (Mikhail Kolmogorov, Jeffrey Yuan, Yu Lin, Pavel. A. Pevzner. Nature Biotechnology (paywalled), bioRxiv
- Flye on GitHub
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#32 Deep tensor factorization and a pitfall for machine learning methods with Jacob Schreiber
29 april 2019 |
75 min
In this episode, we hear from Jacob Schreiber about his algorithm, Avocado.
Avocado uses deep tensor factorization to break a three-dimensional tensor of epigenomic data into three orthogonal dimensions corresponding to cell types, assay types, and genomic loci. Avocado can extract a low-dimensional, information-rich latent representation from the wealth of experimental data from projects like the Roadmap Epigenomics Consortium and ENCODE. This representation allows you to impute genome-wide epigenomics experiments that have not yet been performed.
Jacob also talks about a pitfall he discovered when trying to predict gene expression from a mix of genomic and epigenomic data. As you increase the complexity of a machine learning model, its performance may be increasing for the wrong reason: instead of learning something biologically interesting, your model may simply be memorizing the average gene expression for that gene across your training cell types using the nucleotide sequence.
Links:
- Avocado on GitHub
- Multi-scale deep tensor factorization learns a latent representation of the human epigenome (Jacob Schreiber, Timothy Durham, Jeffrey Bilmes, William Stafford Noble)
- Completing the ENCODE3 compendium yields accurate imputations across a variety of assays and human biosamples (Jacob Schreiber, Jeffrey Bilmes, William Noble)
- A pitfall for machine learning methods aiming to predict across cell types (Jacob Schreiber, Ritambhara Singh, Jeffrey Bilmes, William Stafford Noble)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#31 Bioinformatics Contest 2019 with Alexey Sergushichev and Gennady Korotkevich
24 mars 2019 |
106 min
The third Bioinformatics Contest took place in February 2019.
Alexey Sergushichev, one of the organizers of the contest, and Gennady Korotkevich, the 1st prize winner, join me to discuss this year’s problems.
Timestamps and links for the individual problems:
- Qualification round
- 00:07:14 Bee Population
- 00:14:12 Sequencing Errors
- 00:30:20 Transposable Elements
- Final round
- 00:41:35 Cancer and Chromosome Rearrangements
- 00:56:01 Epigenomic Marks
- 01:10:02 Bacterial Communities
- 01:27:06 Minimal Genome
- 01:34:56 Endangered Species
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#30 Bayesian inference of chromatin structure from Hi-C data with Simeon Carstens
27 februari 2019 |
66 min
Hi-C is a sequencing-based assay that provides information about the 3-dimensional organization of the genome. In this episode, Simeon Carstens explains how he applied the Inferential Structure Determination (ISD) framework to build a 3D model of chromatin and fit that model to Hi-C data using Hamiltonian Monte Carlo and Gibbs sampling.
Links:
- Bayesian inference of chromatin structure ensembles from population Hi-C data (Simeon Carstens, Michael Nilges, Michael Habeck)
- Inferential Structure Determination of Chromosomes from Single-Cell Hi-C Data (Simeon Carstens, Michael Nilges, Michael Habeck)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#29 Haplotype-aware genotyping from long reads with Trevor Pesout
27 januari 2019 |
72 min
Long read sequencing technologies, such as Oxford Nanopore and PacBio, produce reads from thousands to a million base pairs in length, at the cost of the increased error rate. Trevor Pesout describes how he and his colleagues leverage long reads for simultaneous variant calling/genotyping and phasing. This is possible thanks to a clever use of a hidden Markov model, and two different algorithms based on this model are now implemented in the MarginPhase and WhatsHap tools.
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#28 Space-efficient variable-order Markov models with Fabio Cunial
28 december 2018 |
69 min
This time you’ll hear from Fabio Cunial on the topic of Markov models and space-efficient data structures. First we recall what a Markov model is and why variable-order Markov models are an improvement over the standard, fixed-order models. Next we discuss the various data structures and indexes that allowed Fabio and his collaborators to represent these models in a very small space while still keeping the queries efficient. Burrows-Wheeler transform, suffix trees and arrays, tries and suffix link trees, and more!
Links:
- The preprint: A framework for space-efficient variable-order Markov models
- The book: Genome-Scale Algorithm Design
- The GitHub repo
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#27 Classification of CRISPR-induced mutations and CRISPRpic with HoJoon Lee and Seung Woo Cho
29 november 2018 |
57 min
In this episode, HoJoon Lee and Seung Woo Cho explain how to perform a CRISPR experiment and how to analyze its results. HoJoon and Seung Woo developed an algorithm that analyzes sequenced amplicons containing the CRISPR-induced double-strand break site and figures out what exactly happened there (e.g. a deletion, insertion, substitution etc.)
Links:
- CRISPRpic: Fast and precise analysis for CRISPR-induced mutations via prefixed index counting
- CRISPRpic on GitHub
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#26 Feature selection, Relief and STIR with Trang Lê
27 oktober 2018 |
69 min
Relief is a statistical method to perform feature selection. It could be used, for instance, to find genomic loci that correlate with a trait or genes whose expression correlate with a condition. Relief can also be made sensitive to interaction effects (known in genetics as epistasis).
In this episode, Trang Lê joins me to talk about Relief and her version of Relief called STIR (STatistical Inference Relief). While traditional Relief algorithms could only rank features and needed a user-supplied threshold to decide which features to select, Trang’s reformulation of Relief allowed her to compute p-values and make the selection process less arbitrary.
Links:
- Paper: STatistical Inference Relief (STIR) feature selection
- STIR on GitHub
- Relief on Wikipedia
- The original Relief paper by Kira and Rendell (1992)
- Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#25 Transposons and repeats with Kaushik Panda and Keith Slotkin
24 september 2018 |
101 min
Kaushik Panda and Keith Slotkin come on the podcast to educate us about repetitive DNA and transposable elements. We talk LINEs, SINEs, LTRs, and even Sleeping Beauty transposons! Kaushik and Keith explain why repeats matter for your whole-genome analysis and answer listeners’ questions.
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#24 Read correction and Bcool with Antoine Limasset
31 augusti 2018 |
60 min
Antoine Limasset joins me to talk about NGS read correction. Antoine and his colleagues built the read correction tool Bcool based on the de Bruijn graph, and it corrects reads far better than any of the current methods like Bloocoo, Musket, and Lighter.
We discuss why and when read correction is needed, how Bcool works, and why it performs better but slower than k-mer spectrum methods.
Links:
- Preprint: Toward perfect reads: self-correction of short reads via mapping on de Bruijn graphs
- Bcool on GitHub
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#23 RNA design, EteRNA and NEMO with Fernando Portela
27 juli 2018 |
91 min
In this episode, I talk to Fernando Portela, a software engineer and amateur scientist who works on RNA design — the problem of composing an RNA sequence that has a specific secondary structure.
We talk about how Fernando and others compete and collaborate in designing RNA molecules in the online game EteRNA and about Fernando’s new RNA design algorithm, NEMO, which outperforms all prior published methods by a wide margin.
Links:
- The EteRNA game
- The preprint about NEMO
- NEMO project page
- Single-cell RNABIO & organoids meeting in Kiev
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#22 smCounter2: somatic variant calling and UMIs with Chang Xu
29 juni 2018 |
64 min
In this episode I’m joined by Chang Xu. Chang is a senior biostatistician at QIAGEN and an author of smCounter2, a low-frequency somatic variant caller. To distinguish rare somatic mutations from sequencing errors, smCounter2 relies on unique molecular identifiers, or UMIs, which help identify multiple reads resulting from the same physical DNA fragment.
Chang explains what UMIs are, why they are useful, and how smCounter2 and other tools in this space use UMIs to detect low-frequency variants.
Links:
- smCounter2 preprint
- smCounter2 github repository
- smCounter publication
- Review of somatic SNV callers
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#21 Linear mixed models, GWAS, and lme4qtl with Andrey Ziyatdinov
31 maj 2018 |
51 min
Linear mixed models are used to analyze GWAS data and detect QTLs. Andrey Ziyatdinov recently released an R package, lme4qtl, that can be used to formulate and fit these models. In this episode, Andrey and I discuss linear mixed models, genome-wide association studies, and strengths and weaknesses of lme4qtl.
Links:
- Paper: lme4qtl: linear mixed models with flexible covariance structure for genetic studies of related individuals
- lme4qtl on GitHub
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#20 B cell receptor substitution profile prediction and SPURF with Kristian Davidsen and Amrit Dhar
30 april 2018 |
121 min
In this episode Kristian Davidsen and Amrit Dhar present their project called SPURF. SPURF can predict the B cell receptor (BCR) substitution profile of a given clonal family based on a single representative sequence from that family. SPURF works by fitting a tensor regression model to publicly available Rep-seq data.
Links:
- Preprint: Predicting B Cell Receptor Substitution Profiles Using Public Repertoire Data
- Blog post about SPURF by Erick Matsen
- SPURF on GitHub
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#19 Genome fingerprints with Gustavo Glusman
7 april 2018 |
89 min
In this episode, Gustavo Glusman explains his method of reducing a VCF file to a small “fingerprint”, which could be then used to detect duplicate genomes, infer relatedness, map the population structure, and more.
Links:
- The genome fingerprints paper
- The genotype fingerprints preprint
- The data fingerprints preprint
- The blog post about time series visualization
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#18 Bioinformatics Contest 2018 with Alexey Sergushichev and Ekaterina Vyahhi
3 mars 2018 |
113 min
The final round of Bioinformatics Contest 2018 was held on February 24-25th, and the qualification round took place two weeks earlier.
I invited the organizers of the contest, Alexey Sergushichev and Ekaterina Vyahhi, to discuss the problems and find out what it was like to organize the contest.
Timestamps for the problems:
- Qualification round
- 0:41:38 Problem 1. Synthesis of ATP
- 0:48:46 Problem 2. Restriction Sites
- 1:06:42 Problem 3. Tandem Repeats
- Final round
- 1:14:00 Problem 1. Recombination of Plasmids
- 1:25:30 Problem 2. Species Recovering
- 1:28:39 Problem 3. Haplotype Phasing
- 1:36:34 Problem 4. Cluster the Reads
- 1:40:25 Problem 5. Cattle Breeding
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#17 Rarefaction, alpha diversity, and statistics with Amy Willis
22 januari 2018 |
74 min
In this episode, Amy Willis joins me to talk about good and bad ways to estimate taxonomic richness in microbial ecology studies.
Links:
- Rarefaction, alpha diversity, and statistics
- Estimating Diversity via Frequency Ratios
- Estimating the Number of Species in Microbial Diversity Studies
- Summer Institutes 2018 at the University of Washington
- STAMPS: Strategies and Techniques for Analyzing Microbial Population Structures
- bio2040, a new podcast by Flavio Rump
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#16 Javier Quilez on what makes large sequencing projects successful
24 december 2017 |
64 min
Javier Quilez and I discuss what it’s like to be a bioinformatician, how to improve communication between the wet and dry labs and make the research more reproducible.
Make sure to read Javier’s paper we are discussing; it’s a light and entertaining read. The last author on this paper is Guillaume Filion, whom you may remember from the episode on generating functions.
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#15 Optimal transport for single-cell expression data with Geoffrey Schiebinger
26 november 2017 |
69 min
Geoffrey Schiebinger explains how reconstructing developmental trajectories from single-cell RNA-seq data can be reduced to the mathematical problem called optimal transport.
Links:
- Reconstruction of developmental landscapes by optimal-transport analysis of single-cell gene expression sheds light on cellular reprogramming.
- Talk by Geoffrey and Lénaïc Chizat
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#14 Generating functions for read mapping with Guillaume Filion
13 november 2017 |
70 min
Guillaume Filion recently published a preprint in which he applies generating functions, a concept from analytic combinatorics, to estimating the optimal seed length for read mapping.
In this episode, Guillaume and I attempt to explain the core concepts from analytic combinatorics and why they are useful in modeling sequences.
Links:
- Guillaume’s preprint: Analytic combinatorics for bioinformatics I: seeding methods
- Once upon a BLAST
- Guillaume’s blog, «The Grand Locus»
- Dan Gusfield’s home page featuring the fast fourier transform lectures I mention in the podcast
After we recorded the podcast, Guillaume wrote to me to clarify the relationship between read mapping and BLAST:
I looked into my notes about BLAST. The problem that it solves is the following: “Given that a local alignment has score S, what is the probability that it does not contain a word of score T or greater”? The background work of Karlin and Altschul is used to give a statistical significance for S (what is the probability that a “Smith-Waterman random walk” starting at height 0 would reach height S, i.e. what is the probability that aligning two random proteins would yield a score S). The authors write in the original paper “Theory does not yet exist to calculate the probability q that such segment pair will contain a word pair with a score of at least T. However, one argument suggests that q should depend exponentially upon the score of the MSP”.
This is the part that I did not remember well. MSP stands for Maximal Segment Pair, this is the “longest fragment” with “highest score” in the alignment. I thought that Karlin and Altschul solved this part as well, but the authors just go empirical and they calibrate the relationship between T and S with simulations.
I realize a little bit better now that my work is precisely about this problem that the authors of BLAST could not solve, but as you pointed out, I am attacking only a very specific sub-case that is much easier because the models of sequencing error are much simpler than protein evolution. BLAST is concerned with local alignment, so it wants to get all the hits with an MSP score above S. Short read mapping just wants the true location of the read, which does not really have the notion of a score S. But still, mathematically, it is equivalent to the case where S is a constant that depends only on the read size and the distribution of the score T depends only on the seed length and the error rate. I have a few ideas of how to use analytic combinatorics to solve the problem for proteins, but it is mostly complicated because the variable of interest T is a fractional numbers and not an integer…
So what is different from BLAST? The right answer (I think) is that BLAST finds all the hits with an MSP above statistical background, but it says nothing of the probability that the true location contains such an MSP, so it is hard to calibrate the heuristic for that specific problem. In reality, the parallel with BLAST is just the basic strategy: make a statistical model for your problem and use it to calibrate the heuristic.
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#13 Bracken with Jennifer Lu
21 oktober 2017 |
47 min
Jennifer Lu joins me to discuss species abundance estimation from metagenomic sequencing data.
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#12 Modelling the immune system and C-ImmSim with Filippo Castiglione
8 oktober 2017 |
67 min
In this episode, Filippo Castiglione and I discuss different ways to model the immune system.
Links:
- Celada’s and Seiden’s 1992 paper, “A computer model of cellular interactions in the immune system”
- Filippo’s 2001 paper, “Design and implementation of an immune system simulator”
- PLOS One paper, “Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System”
- A paper about modelling sea bass vaccination using C-ImmSim
- C-ImmSim homepage
- The online simulator based on C-ImmSim and the paper describing it
Special thanks to Martina Stoycheva for bringing this work to my attention.
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#11 Collective cell migration with Linus Schumacher
18 september 2017 |
60 min
In this episode, Linus Schumacher joins me to discuss mathematical models of collective cell migration and multidisciplinary research.
Links:
- Semblance of Heterogeneity in Collective Cell Migration and associated code
- Multidisciplinary approaches to understanding collective cell migration in developmental biology
- Linus’s homepage
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#10 Spatially variable genes and SpatialDE with Valentine Svensson
3 september 2017 |
58 min
Valentine Svensson explains how he analyzes spatially-annotated single cell gene expression data using Gaussian processes.
Links:
- Valentine’s preprint, “SpatialDE - Identification Of Spatially Variable Genes”
- SpatialDE code on GitHub
- Valentine’s personal page
- The Integrative Biology & Medicine conference
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#9 Michael Tessler and Christopher Mason on 16S amplicon vs shotgun sequencing
18 augusti 2017 |
46 min
Michael Tessler and Christopher Mason join me to talk about their comparison of 16S amplicon sequencing and shotgun sequencing for quantifying microbial diversity.
Links:
- The 2017 Nature paper that we discuss: Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing
-
Michael’s et al. 2016 paper that describes their original 16S study: A Global eDNA Comparison of Freshwater Bacterioplankton Assemblages Focusing on Large-River Floodplain Lakes of Brazil
-
The sequencing data for these studies is available from NCBI: PRJNA310230 (16S), PRJNA389803 (shotgun)
- Michael’s website
- The Integrative Biology & Medicine conference, featuring the talk on Postmodern Synthesis by Eugene Koonin
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#8 Perfect k-mer hashing in Sailfish
5 augusti 2017 |
22 min
The original version of Sailfish, an RNA-Seq quantification tool, used minimal perfect hash functions to replace k-mers with unique integers. (The current version appears to be using a Cuckoo hashmap instead.)
This is my attempt to explain how a minimal perfect hash function could be built. The algorithm described here is not exactly the same as the one Sailfish used, but it follows the same idea.
Sections:
- Sailfish and perfect hashing (1:15)
- Perfect hashing based on binary search or hash tables (4:34)
- Random hash functions (7:34)
- Perfect hash function based on an acyclic graph (12:16)
Links:
- The Sailfish paper
- The paper describing the perfect hashing algorithm
- The birthday paradox
- Integrative Biology & Medicine
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#7 Metagenomics and Kraken
9 juli 2017 |
28 min
What is metagenomics and how is it different from phylotyping?
What is Kraken and how can it be faster than BLAST?
Let’s try to sort this out.
Sections:
- Culturing and its limitation (00:18)
- Metagenomics vs phylotyping (1:43)
- BLAST (5:53)
- The idea behind Kraken (8:14)
- How Kraken organizes its database (18:08)
Links:
Correction: in this episode, I incorrectly state that Kraken operates on phylogenetic trees, whereas in fact it operates on taxonomic trees.
In practice this means that when Kraken cannot decide among several species, it assigns the read to their lowest common taxon (genus, family etc.), not their latest common evolutionary ancestor.
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#6 Allele-specific expression
25 juni 2017 |
33 min
I talk about allele-specific expression: why it arises and how it can be reliably detected.
Sections:
- The biology of allele-specific expression (2:17)
- Detecting allele-specific expression with RNA-seq (7:46)
- Mapping and sequencing biases (16:39)
- The experiment in yeast (19:47)
- Statistical models (21:44)
Links:
- A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data
- Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data
- The talk by John Marioni
- The blog post explaining the RSEM model
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#5 Relative data analysis and propr with Thom Quinn
10 juni 2017 |
56 min
In this episode, Thom Quinn and I explore different ways to transform and analyze relative data arising in genomics.
We also discuss propr, Thom’s R package to compute various proportionality measures.
Links:
- Thom’s preprint about propr — take a look if you feel lost in all the quantities that we discuss :)
- David Lovell original paper introducing proportionality for relative data and a very detailed appendix
- Ionas Erb’s paper introducing the ρ metric
- Vignettes for propr
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#4 ChIP-seq and GenoGAM with Georg Stricker and Julien Gagneur
29 maj 2017 |
55 min
In this episode, I meet with Georg Stricker and Julien Gagneur from the Technical University of Munich to discuss ChIP-seq data analysis and their tool, GenoGAM.
Links:
- Preprint about GenoGAM
- Georg on GitHub
- Julien’s lab on Twitter
- [BC]2 — The Basel Computational Biology Conference (Basel, September 2017), where you can meet Georg
- The European Human Genetics Conference (Copenhagen, May 2017), where you can meet Daniel Bader from Julien’s lab
Register for the Summer School in Bioinformatics & NGS Data Analysis (#NGSchool2017) (Poland, September 2017)
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#3 miRNA target site prediction and seedVicious with Antonio Marco
12 maj 2017 |
57 min
In this episode Antonio Marco talks about miRNA target site prediction and his tool, seedVicious.
Links:
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#2 Single-cell RNA sequencing with Aleksandra Kolodziejczyk
29 april 2017 |
69 min
In this episode Aleksandra Kolodziejczyk talks about single-cell RNA sequencing.
Links:
- A review paper by Aleksandra
- Comparative analysis of single-cell RNA sequencing methods, including the cost table
- Power Analysis of Single Cell RNA‐Sequencing Experiments
- Monocle: a toolkit for analyzing single-cell gene expression experiments
- Questions from listeners on bioinformatics.chat and reddit
If you enjoyed this episode, please consider supporting the podcast on Patreon.
#1 Transcriptome assembly and Scallop with Mingfu Shao
16 april 2017 |
44 min
In this episode, Mingfu Shao talks about Scallop, an accurate reference-based transcript assembler.
Links:
- The preprint about Scallop
- The preprint about flow decomposition
- The video of a talk by Ben Langmead about Rail-RNA
- The preprint about Rail-RNA
If you enjoyed this episode, please consider supporting the podcast on Patreon.
00:00
-00:00
En liten tjänst av I'm With Friends. Finns även på engelska.